ga4gh_challenge
Workflow for GA4GH/DREAM challenge (phase 2)
Project maintained by Barski-lab Hosted on GitHub Pages — Theme by mattgraham
![]()
ChIP-Seq-SE CWL pipeline from BioWardrobe
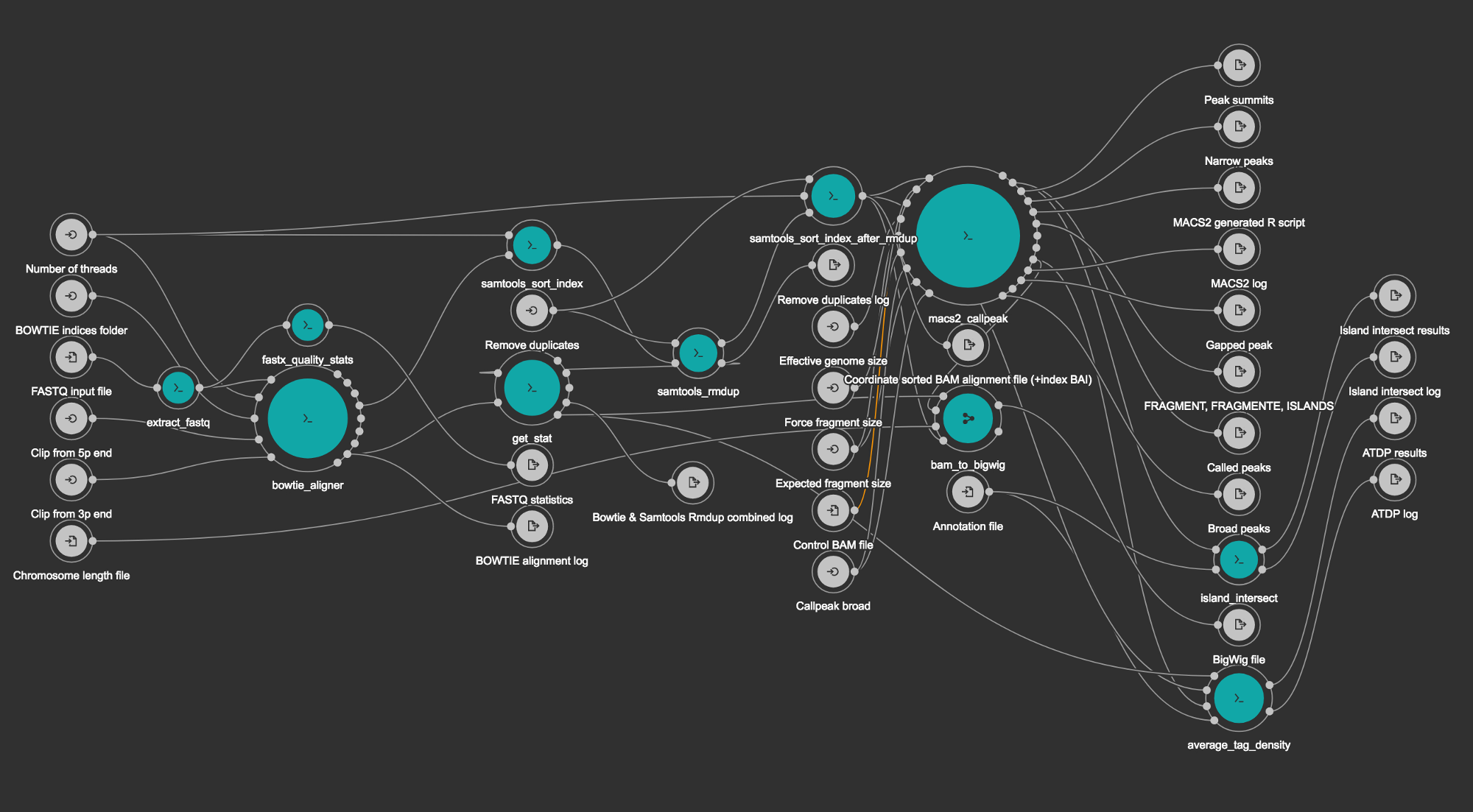
This workflow is a CWL version of a Python pipeline from BioWardrobe (Kartashov and Barski, 2015). It starts by extracting input FASTQ file (if it was compressed). Next step runs BowTie (Langmead et al., 2009) to perform alignment to a reference genome, resulting in an unsorted SAM file. The SAM file is then sorted and indexed with Samtools (Li et al., 2009) to obtain a BAM file and a BAI index. Next MACS2 (Zhang et al., 2008) is used to call peaks and to estimate fragment size. In the last few steps, the coverage by estimated fragments is calculated from the BAM file and is reported in bigWig format. The pipeline also reports statistics, such as read quality, peak number and base frequency, and other troubleshooting information using tools such as Fastx-toolkit and Bamtools.
This workflow v0.0.2 was used for GA4GH-DREAM Workflow Execution Challenge.
Requirements
- Ubuntu 16.04.4 or macOS 10.13.5
- Either of Python 2.7, 3.5, 3.6 or 3.7
- docker (for Ubuntu read here, for macOS read here)
- pip (link to the latest stable pip)
- cwltool (install from here)
To run the workflow
- Download the latest workflow version
$ git clone --recursive https://github.com/Barski-lab/ga4gh_challenge.gitUse
--recursiveto fetch the ga4gh_challenge_data submodule (download time depends on your Internet connection speed) - Prepare input data
$ cd ./ga4gh_challenge/data $ ./prepare_inputs.shThe script will uncompress the input FASTQ file.
- Run the workflow (tested with cwltool==1.0.20181217162649)
$ mkdir ../outputs && cd ../outputs $ cwltool ../biowardrobe_chipseq_se.cwl ../biowardrobe_chipseq_se.yamlResults will be saved into the
outputsdirectory ___ View the workflow in CWLviewer
Pipeline’s structural scheme generated by Rabix Composer.